

Chapter 1: Introduction to Drug-Device Combination Products
1.1 Understanding Combination Products
In today's pharmaceutical landscape, nearly half of the top 20 drugs[1] incorporate both a drug constituent and at least one device constituent. These device components are typically used for drug preparation, reconstitution, or delivery. Products that combine drugs with devices are commonly referred to as Combination Products, as they bring together two or more regulated product types. While the term "Combination Product" is widely used in the industry, it is currently defined in only a few jurisdictions.
1.2 Types of Combination Products
The most comprehensive definition of Combination Products can be found in the Code of Federal Regulations (CFR) Title 21 (Part 3.2)[2]. According to this definition, Combination Products encompass four types of products that require the combination of at least two regulated components. These regulated components include Drugs, Devices, and Biological products. A Combination Product always involves the combination of different regulations, such as Drugs and Devices, Biologics and Devices, or Drugs and Biologics. For the purpose of this article, Drugs and Biological products will be treated interchangeably, with any reference to Drugs also including Biological products. Additionally, this article focuses specifically on Combination Products that include devices only. Therefore, Combination Products consisting solely of Drugs and Biological products are not within the scope, nor are Combination Products that consist of devices with an ancillary drug component.
In the United States, the first type of Combination Product is referred to as the "single-entity Combination Product." These products involve the physical, chemical, or other combination or mixing of two or more regulated components, resulting in a single entity. Examples of single-entity Combination Products include pre-filled syringes and single-use pens.
The second type, known as "co-packaged Combination Products," involves packaging at least one device together with a drug in a single package or unit. This category encompasses a wide range of products, such as cannulas, vial adapters, measuring spoons, inhalation devices, and multiple-use pens.
The remaining two types of Combination Products fall under the category of "cross-labeled Combination Products." In these cases, the different components are not packaged together, but the labeling or investigational plan of one component (e.g., drug) refers to another component (e.g., device), and both components are necessary to achieve the intended use, indication, or effect. The regulation distinguishes between cases where both components are still in the investigational stage and cases where one component has already been approved.
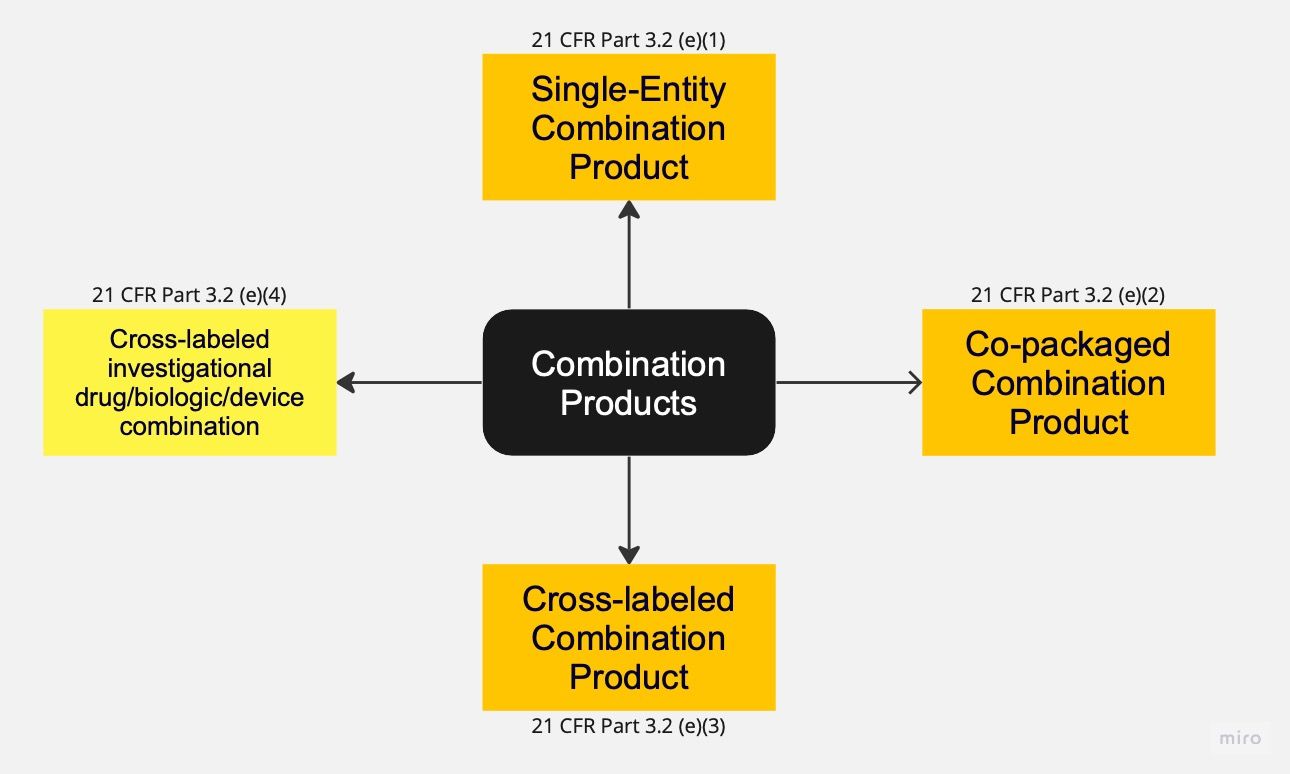
An example of a cross-labeled Combination Product with both components in the investigational stage is a new inhalation drug where the inhalation powder capsules are not packed together with the dry powder inhaler, as they are distributed separately.
In situations where one component has already been approved, the combination of the two components would be considered a Combination Product only if the approval of the proposed product requires a change in the labeling of the approved product to reflect a change in intended use, dosage, form, strength, route of administration, or a significant change in dose.
While this comprehensive definition is currently applicable only in the United States, the categorizations of "single-entity," "co-packaged," and "cross-labeled" Combination Products can be used globally. However, it should be noted that in practice, clear differentiation between these types is not always possible. Some Combination Products may combine two or more of these types. For example, a Combination Product may consist of a drug in a pre-filled syringe, a co-packaged injection needle, and a separately distributed multiple-use auto-injector, encompassing all three types within a single product.
By understanding the various types of Combination Products, we can delve deeper into the regulations and implications surrounding these innovative healthcare solutions.
Chapter 2: The Purpose of Combination Product Regulations
The regulation of Combination Products, as established in the United States, serves two main purposes.
Firstly, it allows for the consolidation of market authorizations for all components of a Combination Product under a single regulatory pathway. Prior to the introduction of this regulation in the US, the device components of Combination Products obtained market authorization through device clearance or approval pathways, such as Pre-market Notification 510(k) or Premarket Approval (PMA). On the other hand, the drug components were independently approved through drug approval pathways, such as New Drug Application (NDA). Today, all components of a Combination Product can be approved under one pathway, based on the "primary mode of action" (PMOA) of the Combination Product. For instance, a drug combined with its respective drug-delivery device would receive market authorization through a New Drug Application (NDA), while a Biological product would be approved under a Biologics License Application (BLA).
The second purpose of Combination Product regulation is to establish a holistic approach to their review. This means that Combination Products are evaluated as a whole, rather than as isolated components. The underlying premise is that a Combination Product offers more than just the sum of its individual benefits and risks. When an approved drug is combined with an approved device, this combination introduces additional benefits as well as potential changes in risks. Therefore, the review process must be conducted on an individual basis, taking into account the specific characteristics and risks associated with each Combination Product. For example, the severity of risks associated with a drug-delivery device depends on the specific drug it is intended to deliver.
The holistic approach is not only reflected in the consolidation of marketing authorizations but also in additional quality and regulatory requirements for Combination Products. During the development process, Design Controls according to CFR Title 21 Part 820.30[3] are expected to be applied not only to the device component but also to the Combination Product as a whole. Usability engineering is another aspect that is expected to be considered for Combination Products.
By adopting this holistic approach, regulators aim to ensure that Combination Products meet the necessary quality and safety standards, taking into account the unique characteristics and interactions between the drug and device components.
In summary, the regulation of Combination Products in the United States serves the purpose of streamlining market authorizations and implementing a comprehensive approach that considers the combined benefits and risks of these innovative healthcare solutions.
Chapter 3: Regulation of Combination Products
Drug-Device Combination Products can be marketed in various jurisdictions, although specific regulations for Combination Products are currently only available in the United States and Japan. In other countries, the rules primarily address single-entity Combination Products. Nonetheless, it is crucial to understand how the marketing of Drug-Device Combination Products is possible in major markets.
3.1 Regulation in the United States
In the United States, Combination Products are subject to specific regulations. Applicants for Combination Products obtain marketing authorization through the pathway determined by the constituent part that provides the Primary Mode of Action (PMOA). In cases where there is uncertainty regarding the premarket review procedure, the US Food and Drug Administration (FDA) offers support to applicants through a Request for Designation (RFD) process.
While the definitions for Combination Products are provided in CFR Title 21 Part 3.2, there is an additional Regulation of Combination Products codified in Part 4[4]. This regulation consists of subpart A, "Current Good Manufacturing Practice Requirements for Combination Products," and subpart B, "Postmarketing Safety Reporting for Combination Products."
Subpart A establishes the current good manufacturing practice (CGMP) requirements applicable to Combination Products. For Combination Products covered by this article, three separate chapters of the CFR are relevant. Device CGMPs are described in CFR Title 21 Part 820[5], Drug CGMPs are included in CFR Title 21 Parts 210/211[6], and CGMPs for biological products are summarized in CFR Title 21 Parts 600–680[7].
When a Combination Product combines products from different chapters, it is necessary to identify the applicable CGMPs. Manufacturers have the option to comply with all CGMPs relevant to the respective constituent parts. Compliance with all CGMPs is mandatory if constituent parts are manufactured at different facilities, even if they belong to the same manufacturer.
For manufacturers producing single-entity or co-packaged Combination Products in a single facility, subpart A provides options to use only one CGMP, with specific components of the other CGMPs amended accordingly. If a manufacturer has established a quality management system in compliance with the drug CGMPs, the Combination Product can be manufactured under this system, provided additional requirements from the device CGMPs are fulfilled. Similarly, if a manufacturer has established device CGMPs, those would be considered sufficient, with additional requirements from the drug CGMPs needing to be met. For Combination Products containing a biological product constituent part, compliance with all requirements laid down in Parts 600–680 is necessary.
Subpart B of CFR Title 21 Part 4 establishes requirements for Postmarketing Safety Reporting for Combination Products. Manufacturers of Drug-Device Combination Products are required to perform vigilance reporting according to both drug-reporting and device-reporting regulations. Any Adverse Drug Reactions or Malfunctions related to the Combination Product must be attributed to the respective part (Drug or Device) and reported accordingly.
3.2 Regulation in the European Union (EU)
In the EU, the regulatory landscape for medical devices has undergone changes. Nowadays, medical devices are governed by the Medical Device Regulation 2017/745 (MDR)[8], which has replaced the Medical Device Directive 93/42/EEC (MDD).
Specific regulations for Combination Products do not exist in the EU. Drug-Device combinations are regulated either under device regulations, drug regulations, or both.
The MDR recognizes two types of single-entity Combination Products in the EU. The first type includes devices intended to administer a medicinal product, forming a single integral product exclusively for use in that combination and not reusable (as per the second subparagraph of Article 1(9) MDR), as well as devices incorporating, as an integral part, a substance which, if used separately, would be considered to be a medicinal product while the action of that substance is principal and not ancillary to that of the device (as per the second subparagraph of Article 1(8) MDR). Such products are governed by the drug regulations outlined in the Medicinal Product Directive 2001/83/EC (MPD). However, the MDR requires compliance with the relevant General Safety and Performance Requirements (GSPR) as per Annex I for the device. As part of the Marketing Authorization Dossier for such a single-integral product, an opinion of a Notified Body needs to be included, as described in Article 117 of the MDR. This Notified Body Opinion (NBOp) is the result of a procedure initiated by the applicant where a Notified Body confirms whether or not the device part is in compliance with the General Safety and Performance Requirements (GSPR). A Notified Body Opinion is required for new Marketing Authorization Applications as well as Variations related to substantial changes to the device part of already approved products.
The second type described in the MDR involves devices containing a substance considered a medicinal product, including medicinal products derived from human blood or human plasma, where the substance provides an action ancillary to that of the device. These products, such as certain drug-coated stents, are regulated as devices under the MDR and are not within the scope of this article.
The Medical Device Coordination Group (MDCG) Document MDCG 2022-5[9] provides a summary and interpretation of these requirements. Further guidance is provided by the EMA in the Q&A documen[10] for the implementation of the MDR.
In the EU, co-packaged and cross-labeled Combination Products are not explicitly regulated. Instead, the device constituent parts of such Combination Products are regulated under the MDR, requiring conformity assessment procedures and the issuance of a CE mark. From a drug perspective, the marketing authorization dossier is expected to include details of the devices used or administered with the medicinal product.
Regarding quality requirements, current regulations necessitate that pharmaceutical companies consider the quality requirements for the device component of any single-entity Combination Product. Most of these provisions are directly or indirectly outlined in the essential requirements described in Annex I of the MDR. They include Risk Management, Usability Engineering, and the use of Design Controls.
With regard to Dossier requirements, the European Medicines Agency (EMA) published the Scientific guideline on "Quality documentation for medicinal products when used with a medical device,"[11] which describes the information that should be presented in the quality part of a marketing authorization dossier for a medicinal product when it is used with a medical device. The guideline focuses on product-specific quality aspects of a medical device (integral, co-packaged, or separately-obtained and referenced in the product information) that may have an impact on the quality, safety, and efficacy of a medicinal product.
3.3 Regulation in Japan
In Japan, the Notification (PFSB/ELD Notification No. 1024-2) on the "Handling of Marketing Application for Combination Products"[12] was published on October 24, 2014. This notification provides a definition for Combination Products, including three types of products: single-entity, co-packaged, and cross-labeled Combination Products. However, cross-labeled Combination Products are not recognized in Japan. The notification governs marketing applications for Combination Products, similar to the requirements in the United States. Combination Products are treated as a single product under drug, device, or cellular/tissue-based product regulations, depending on their characteristics.
Similar to the United States, since November 24, 2016, marketing authorization holders of Combination Products in Japan are required to perform Postmarketing Safety Reporting in accordance with the requirements for the constituent part.
From a quality perspective, there are no specific quality systems requirements for Combination Products in Japan. The requirements applied to medical devices also apply to the device constituent parts of Combination Products.
In conclusion, the regulation of Combination Products varies across different jurisdictions. The United States has well-established regulations for Combination Products, considering both individual components and the Combination Product as a whole. In the EU, single-entity Combination Products are recognized, and new regulations, such as Article 117 of the MDR, will impact the conformity assessment process. Japan has specific regulations governing Combination Products, while other countries primarily address single-entity Combination Products. It is essential for manufacturers and stakeholders to navigate these regulatory landscapes to ensure compliance and successful market authorization of Drug-Device Combination Products.
Chapter 4: Conclusion
The regulatory and quality requirements for Drug-Device Combination Products vary from country to country. Understanding these requirements is crucial for manufacturers and stakeholders involved in the development and marketing of these innovative healthcare solutions.
In the United States, Combination Products are subject to specific regulations that aim to streamline market authorizations and establish a comprehensive approach to their review. The consolidation of market authorizations under one pathway based on the primary mode of action (PMOA) of the Combination Product allows for a more efficient regulatory process. The holistic approach considers the combined benefits and risks of the drug and device components, ensuring that the unique characteristics and interactions are adequately evaluated.
In the European Union (EU), specific regulations for Combination Products are currently limited. Single-entity Combination Products are recognized, and the upcoming Medical Device Regulation (MDR) will introduce new requirements, such as Article 117, which will impact the conformity assessment process. The EU is also working on developing guidelines to address the quality requirements of Combination Products, which will likely increase the content requirements for marketing authorization dossiers.
Japan has its own regulations governing Combination Products, providing clarity on marketing applications and postmarketing safety reporting. However, specific quality systems requirements for Combination Products are not outlined in Japanese regulations.
As the field of Combination Products continues to evolve, it is essential for manufacturers to stay informed about the regulatory landscape in different jurisdictions. Compliance with the relevant regulations and adherence to quality requirements are crucial for successful market authorization and ensuring the safety and efficacy of these innovative healthcare solutions.
In conclusion, navigating the regulatory and quality requirements for Drug-Device Combination Products is a complex task. Collaboration between regulatory authorities, manufacturers, and stakeholders is essential to ensure the development and availability of safe and effective Combination Products that can improve patient outcomes and advance healthcare innovation.
”The 50 best-selling pharmaceuticals of 2022: COVID-19 vaccines poised to take a step back”, by Brian Buntz, Drug Discovery & Development ↩︎
MDCG 2022 – 5 - “Guidance on borderline between medical devices and medicinal products under Regulation (EU) 2017/745 on medical devices” ↩︎
EMA/37991/2019 - EMA Questions & Answers for applicants, marketing authorisation holders of medicinal products and notified bodies with respect to the implementation of the Medical Devices and In Vitro Diagnostic Medical Devices Regulations ((EU) 2017/745 and (EU) 2017/746) ↩︎
EMA/CHMP/QWP/BWP/259165/2019 - EMA Guideline on quality documentation for medicinal products when used with a medical device ↩︎
Japan Ministry of Health, Labour and Welfare/PMDA Notification PFSB/ELD Notification No. 1024-2 on Handling of Marketing Application for Combination Products ↩︎